



















What our community say
Featured Publications
Best-in-Class Performance in cfDNA Isolation: Side by Side Comparison
Results and discussion
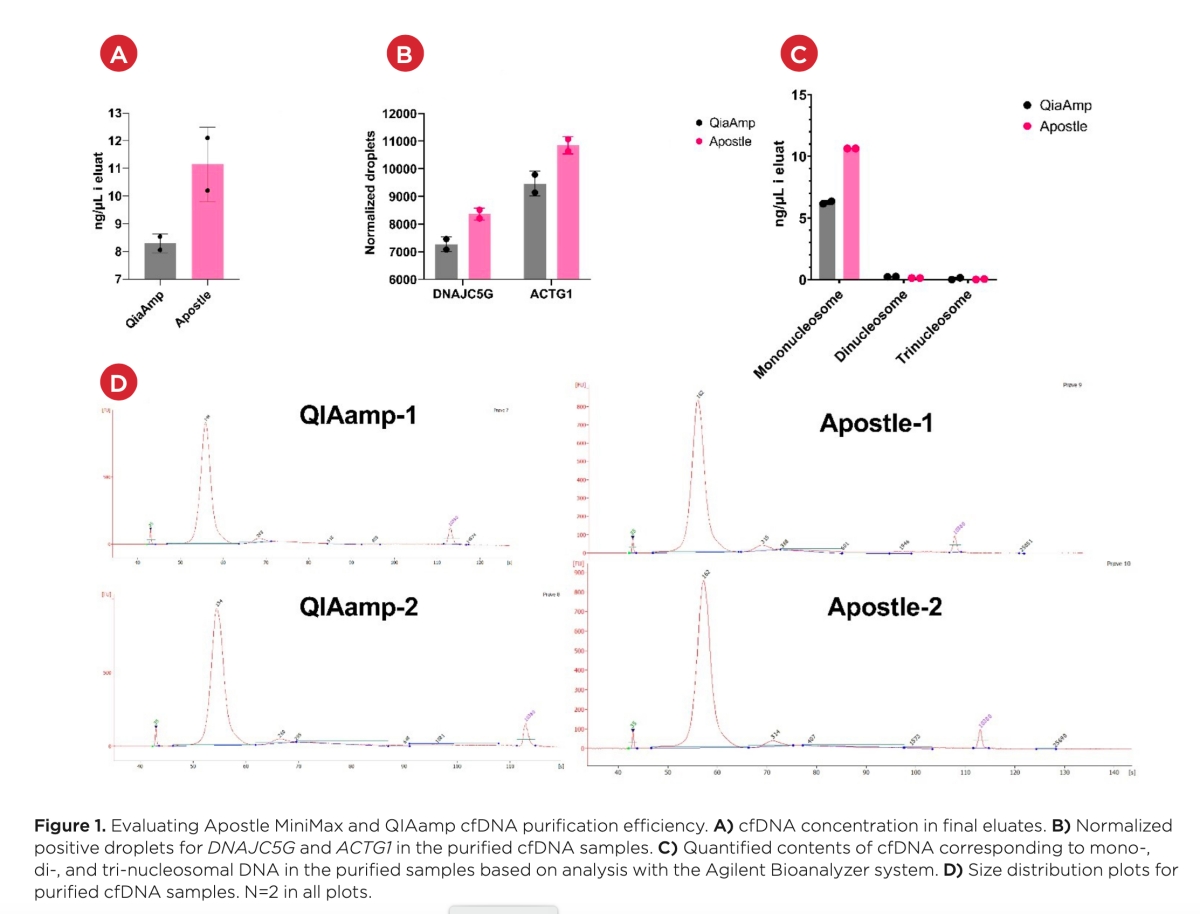
Mean cfDNA concentrations in the eluates were determined to be 8.30 ng/μL for samples extracted with the QIAamp kit and 11.15 ng/μL for samples extracted with the Apostle kit when measured with the Qubit assay (Figure 1A). This illustrates a 34.3% higher yield when using the Apostle kit as compared to the QIAamp kit.
When evaluating the contents of specific genes in the eluate, the Apostle purification displayed a higher yield than QIAamp (Figure 1B). 8358 DNAJC5G positive droplets were detected in the Apostle samples compared to 7269 droplets in the QIAamp samples. Similarly, 10849 ACTG1 droplets were detected in the Apostle-extracted samples versus 9463 droplets detected in the QIAamp-extracted samples. This data demonstrates an average of 14.8% higher yield using the Apostle kit.
Finally, when evaluating cfDNA fragments via the Agilent bioanalyzer instrument, the Apostle kit demonstrated the highest yield (Figure 1C and D). Mononucleosomal cfDNA concentration in the eluate of Apostle samples was estimated to be 10.64 ng/μL versus 6.26 ng/μL in the eluate of QIAamp samples. The dinucleosomal cfDNA concentration was low for both kits, with 0.13 ng/μL recorded for Apostle samples and 0.24 ng/μL for QIAamp samples. This indicates a slightly better purification of dinucleosomal cfDNA using QIAamp; however, this could also be an artifact given the low concentrations. The Apostle mononucleosomal cfDNA yield was 70.0% higher than the yield for the QIAamp samples.
Publication: Comparative analysis of cell-free DNA extraction efficiency from plasma. Beckman Coulter Life Sciences. Indianapolis, IN. Technical Note 2022.
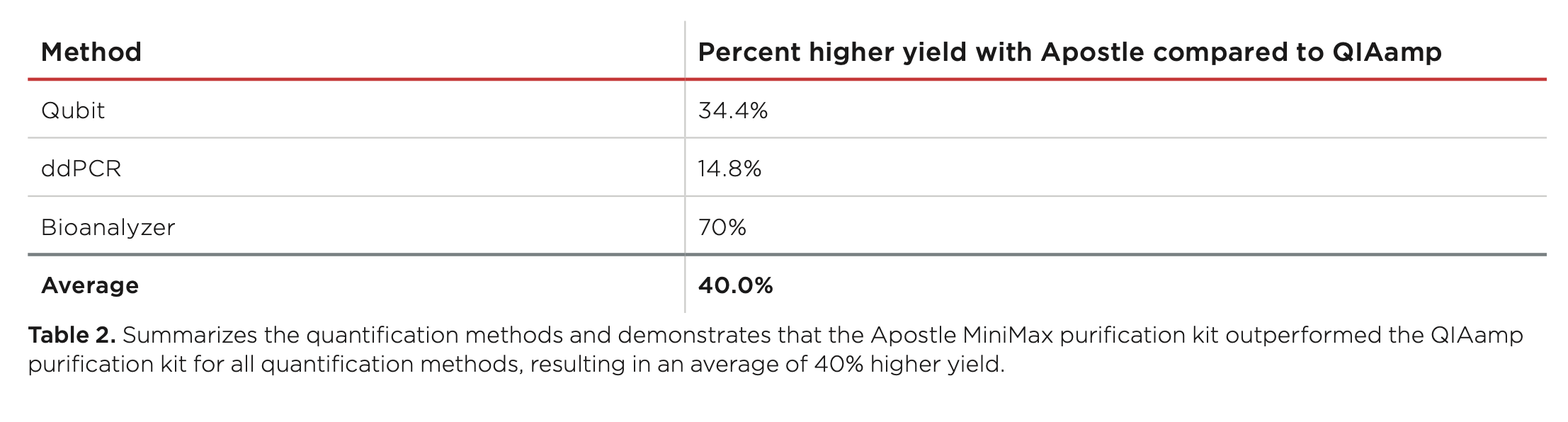
A summary provided in Table 2 shows the consistently higher yield achieved by the Apostle MiniMax High Efficiency cfDNA Isolation Kit as compared to the QIAamp® circulating nucleic acid kit. The average 40% improvement in yield underscores the importance of extraction kit selection in cfDNA assay development and highlights the potential impact of cfDNA isolation efficiency in ctDNA detection.
Publication: Comparative analysis of cell-free DNA extraction efficiency from plasma. Beckman Coulter Life Sciences. Indianapolis, IN. Technical Note 2022.
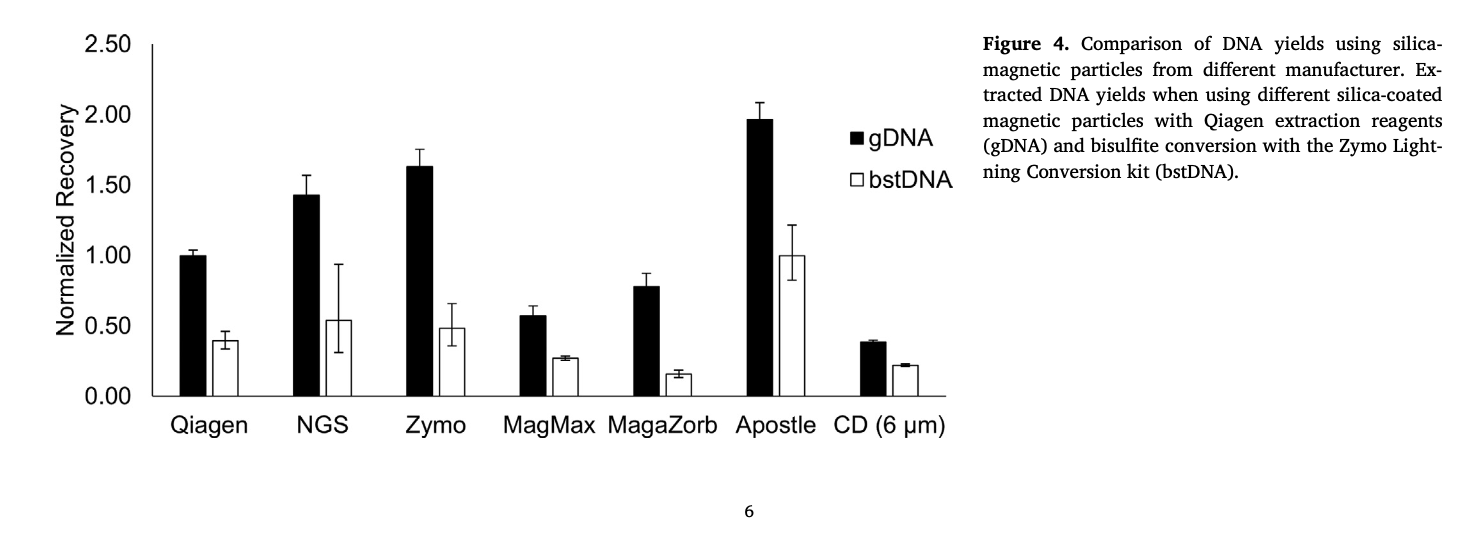
(Figure 4) - "Most notably, the Apostle particles outperformed all others, achieving almost 2-fold higher recovery yields than the particles supplied in the Qiagen kit. "
Publication: High-throughput sample processing for methylation analysis in an automated, enclosed environment. Stark et al. SLAS Technology 2022; 27(3):172-179.

Publication: Optimization of high-volume cell free DNA extraction and end-to-end automation for TSO 500 ctDNA library prep [abstract]. Nripesh Prasad, Rachel Rock, Rebecca Beatty, Melanie Robinson, Dineen Wildman, Michael Sykes, Boris Umylny, Thomas Halsey. In: Proceedings of the American Association for Cancer Research Annual Meeting 2024; Part 1 (Regular Abstracts); 2024 Apr 5-10; San Diego, CA. Philadelphia (PA): AACR; Cancer Res 2024;84(6_Suppl):Abstract nr 2298.
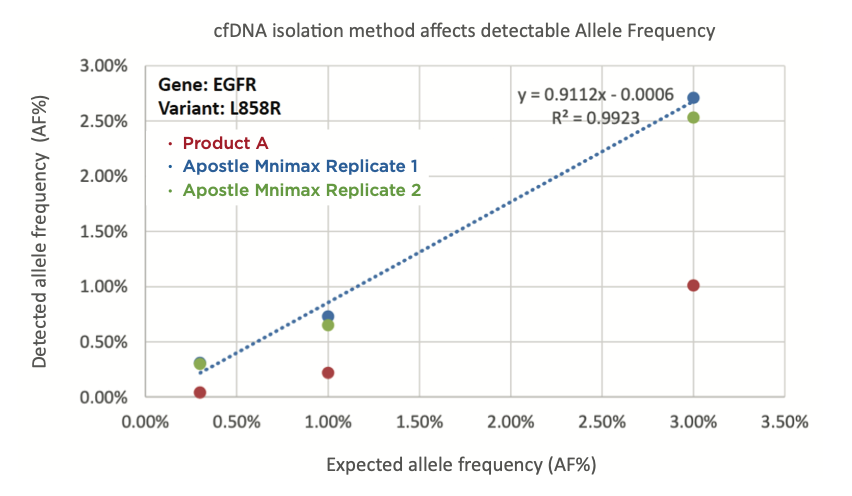
Figure 1. cfDNA extraction method affects allele frequency (AF%). EGFR L858R standards with AF% of 0.3%, 1.0%, and 3.0% were spiked into plasma and isolated using Product A (red) or Apostle MiniMax cfDNA isolation kit (blue and green). The eluates were analyzed by NGS (performed by the Institute B) following standard protocol and AF% calculated using their established workflow. cfDNA isolated by Apostle MiniMax cfDNA isolation kit shows higher concordance between detected AF% and expected AF%.
Publication: cfDNA Extraction Efficiency Affects NGS Data. Beckman Coulter Life Sciences. Indianapolis, IN. Technical Note 2020.
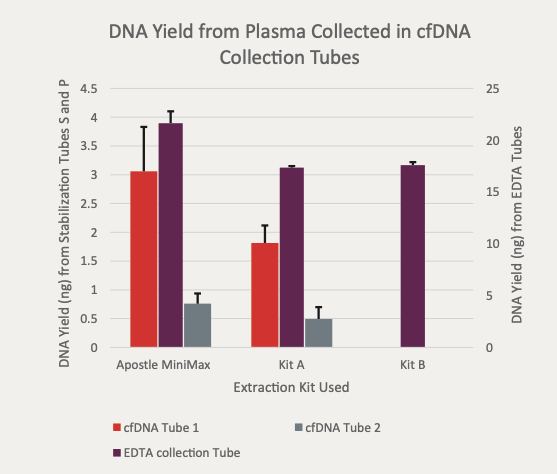
(Figure 1) "For each tube the Apostle MiniMax extracted higher total yield of DNA. "
Publication: cfDNA Extraction from Plasma for Liquid Biopsy: Apostle MiniMaxTM High Efficiency cfDNA Isolation Kit. Beckman Coulter Life Sciences, Data Sheet. 2019.
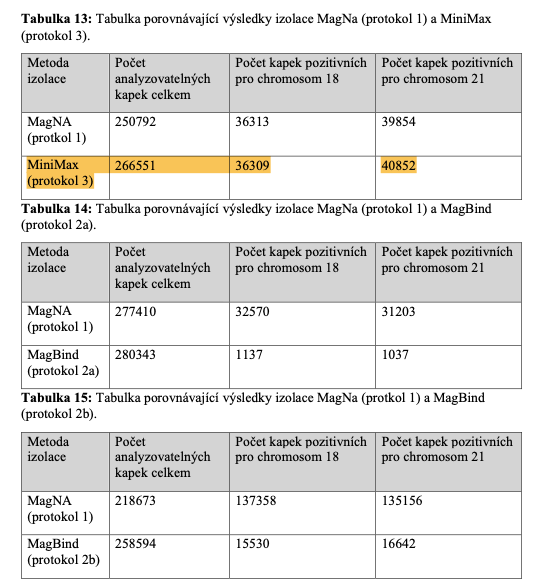
(Note: Table 13 referenced below in this diploma thesis shows that Apostle MiniMax yields the greatest number of analyzable drops in the digital PCR.)
Publication. Optimalizace digitální polymerázové řetězové reakce pro aplikaci v neinvazivní prenatální diagnostice (Optimization of digital polymerase chain reaction for application in non-invasive prenatal diagnostics) Author: Šenkyřík, Pavel; Advisor: Korabečná, Marie; Referee: Vodička, Radek; Faculty / Institute: Faculty of Science; Discipline: Anthropology and Human Genetics; Department: Department of Anthropology and Human Genetics; Date of defense: 13. 9. 2022; Publisher: Univerzita Karlova, Přírodovědecká fakulta; Language: Czech






























































Publications (2024)
Publications (2023)
Publications (2022)
42. Comparative analysis of cell-free DNA extraction efficiency from plasma. Beckman Coulter Life Sciences. Indianapolis, IN. Technical Note 2022.
(Note: Apostle MiniMax technology is compared and discussed in this study.)
Results and discussion
Mean cfDNA concentrations in the eluates were determined to be 8.30 ng/μL for samples extracted with the QIAamp kit and 11.15 ng/μL for samples extracted with the Apostle kit when measured with the Qubit assay (Figure 1A). This illustrates a 34.3% higher yield when using the Apostle kit as compared to the QIAamp kit.
When evaluating the contents of specific genes in the eluate, the Apostle purification displayed a higher yield than QIAamp (Figure 1B). 8358 DNAJC5G positive droplets were detected in the Apostle samples compared to 7269 droplets in the QIAamp samples. Similarly, 10849 ACTG1 droplets were detected in the Apostle-extracted samples versus 9463 droplets detected in the QIAamp-extracted samples. This data demonstrates an average of 14.8% higher yield using the Apostle kit.
Finally, when evaluating cfDNA fragments via the Agilent bioanalyzer instrument, the Apostle kit demonstrated the highest yield (Figure 1C and D). Mononucleosomal cfDNA concentration in the eluate of Apostle samples was estimated to be 10.64 ng/μL versus 6.26 ng/μL in the eluate of QIAamp samples. The dinucleosomal cfDNA concentration was low for both kits, with 0.13 ng/μL recorded for Apostle samples and 0.24 ng/μL for QIAamp samples. This indicates a slightly better purification of dinucleosomal cfDNA using QIAamp; however, this could also be an artifact given the low concentrations. The Apostle mononucleosomal cfDNA yield was 70.0% higher than the yield for the QIAamp samples.
A summary provided in Table 2 shows the consistently higher yield achieved by the Apostle MiniMax High Efficiency cfDNA Isolation Kit as compared to the QIAamp® circulating nucleic acid kit. The average 40% improvement in yield underscores the importance of extraction kit selection in cfDNA assay development and highlights the potential impact of cfDNA isolation efficiency in ctDNA detection.
36. High-throughput sample processing for methylation analysis in an automated, enclosed environment. Alejandro Stark, Thomas R. Pisanic, James G. Herman, Tza-Huei Wang. SLAS Technology Volume 27, Issue 3, June 2022, Pages 172-179.
(Note: Apostle MiniMax technology is compared and discussed in this study.)
(Figure 4) - "Most notably, the Apostle particles outperformed all others, achieving almost 2-fold higher recovery yields than the particles supplied in the X kit. "
Variation in methylcytosine is perhaps the most well-studied epigenetic mechanism of gene regulation. Methods that have been developed and implemented for assessing DNA methylation require sample DNA to be extracted, purified and chemically-processed through bisulfite conversion before downstream analysis. While some automated solutions exist for each of these individual process steps, a fully integrated solution for accomplishing the entire process in a high-throughput manner has yet to be demonstrated. Thus, sample processing methods still require numerous manual steps that may reduce sample throughput and precision, while increasing the risk of contamination and human error. In this work, we present an integrated, automated solution for performing the entire sample preparation process, including DNA extraction, purification, bisulfite conversion and PCR plate preparation within in an enclosed environment. The method employs silica-coated magnetic particles that eliminate the need for a centrifuge or vacuum manifold, thereby reducing the complexity and cost of the required automation platform. Toward this end, we also compare commercial DNA extraction and bisulfite conversion kits to identify a protocol suitable for automation to significantly improve genomic and bisulfite-treated DNA yields over manufacturer protocols. Overall, this research demonstrated development of an automated protocol that offers the ability to generate high-quality, bisulfite-treated DNA samples in a high-throughput and clean environment with minimal user intervention and comparable yields to manual processing.
34. Optimalizace digitální polymerázové řetězové reakce pro aplikaci v neinvazivní prenatální diagnostice (Optimization of digital polymerase chain reaction for application in non-invasive prenatal diagnostics) Author: Šenkyřík, Pavel; Advisor: Korabečná, Marie; Referee: Vodička, Radek; Faculty / Institute: Faculty of Science; Discipline: Anthropology and Human Genetics; Department: Department of Anthropology and Human Genetics; Date of defense: 13. 9. 2022; Publisher: Univerzita Karlova, Přírodovědecká fakulta; Language: Czech
(Note: Apostle MiniMax technology is discussed in this diploma thesis.)
(Note: Table 13 referenced below in this diploma thesis shows that Apostle MiniMax yields the greatest number of analyzable drops in the digital PCR.)
Publications (2021)
Publications (2020)
13. cfDNA Extraction Efficiency Affects NGS Data. Han Wei, Ph.D., Beckman Coulter Life Sciences. Indianapolis, IN. Technical Note 2020.
Figure 1. cfDNA extraction method affects allele frequency (AF%). EGFR L858R standards with AF% of 0.3%, 1.0%, and 3.0% were spiked into plasma and isolated using Product A (red) or Apostle MiniMax cfDNA isolation kit (blue and green). The eluates were analyzed by NGS (performed by the Institute B) following standard protocol and AF% calculated using their established workflow. cfDNA isolated by Apostle MiniMax cfDNA isolation kit shows higher concordance between detected AF% and expected AF%.
In order to understand how cfDNA extraction efficiency affects NGS data, we present Institute B titration data. EGFR L858R standards with an allele frequency (AF%) of 0.3%, 1.0%, and 3.0% were spiked into plasma and isolated using either Apostle MiniMaxTM High Efficiency cfDNA Isolation or Product A. The cfDNA was analyzed via NGS and AF% was calculated by Institute B. cfDNA isolated by Apostle MiniMax cfDNA isolation kit shows concordance between detected AF% and expected AF%.
For cancer samples, the allele frequency represents the percentage of sequence reads carrying a mutant allele of an individual patient’s cancer. Researchers often set thresholds for defining the variant allele frequency as mutation; therefore, low cfDNA recovery efficiency can decrease sensitivity and lead to an increase in false negatives for mutation calling.
Publications (2019)
2. cfDNA Extraction from Plasma for Liquid Biopsy: Apostle MiniMaxTM High Efficiency cfDNA Isolation Kit. Beckman Coulter Life Sciences, Data Sheet. 2019.
(Figure 1) "For each tube the Apostle MiniMax extracted higher total yield of DNA. "
Apostle MiniMax High Efficiency cfDNA Isolation Kit, Apostle MiniMax is a cell–free DNA (cfDNA) isolation reagent kit, built on magnetic bead–based technology. Apostle MiniMax has been demonstrated to purify cfDNA from human plasma in both manual and automated workflows.
• Data representative of results of cfDNA extracted from 1–5mL of plasma
• Demonstrated compatibility with a variety of collection tubes
• cfDNA purity shown to be suitable for downstream PCR based assays
Publications (2018)































































































Team's track record of publications





